

ADSS1ミオパチーについて
ADSS1ミオパチーの症状

幼児期の成長発達は正常に経過します。
小児期では極端に足が遅く疲れやすい特徴がありますが、日常生活動作には困難さの自覚がない為、病気に気づく事は思春期までは殆どありません。
思春期に階段が登りづらい、思うように走れないなどで違和感を感じるようになります。
発症時に著明な握力低下が指摘され、筋力低下の初期症状の可能性があり、握力低下は特徴的な徴候のひとつです。
筋力低下以外にも呼吸機能障害、嚥下障害、心臓の異常などを併発することがあり、症状は多岐にわたります。
進行していくと歩行の困難さに加え、嚥下障害による経管栄養と胃ろう造設、呼吸機能障害による人工呼吸器のサポートが必要となる人もいます。心臓に関しては25%の方に左室肥大が認められます。
人によって出てくる症状は様々であり、歩行が困難でも呼吸機能、心機能に影響がない方もいます。また、若いうちから様々な症状が出てくる方もいます。
ADSS1ミオパチーの診断について
ADSS1ミオパチーは過去に遠位型ミオパチーとして報告されてますが、全症例にネマリン小体が認められることからネマリンミオパチーとされています。
ネマリンミオパチーの約30%がADSS1遺伝子が原因とされています。
遺伝子検査をされていない場合、ネマリンミオパチーとして先天性ミオパチーとするか、遠位型ミオパチーとするかは現在主治医の判断により異なっている可能性があります。
実はADSS1ミオパチーであっても必ずしも先天性ミオパチーとして難病指定を受けているとはいえない状況です。
ADSS1ミオパチーは常染色体 潜性遺伝(劣性遺伝)です
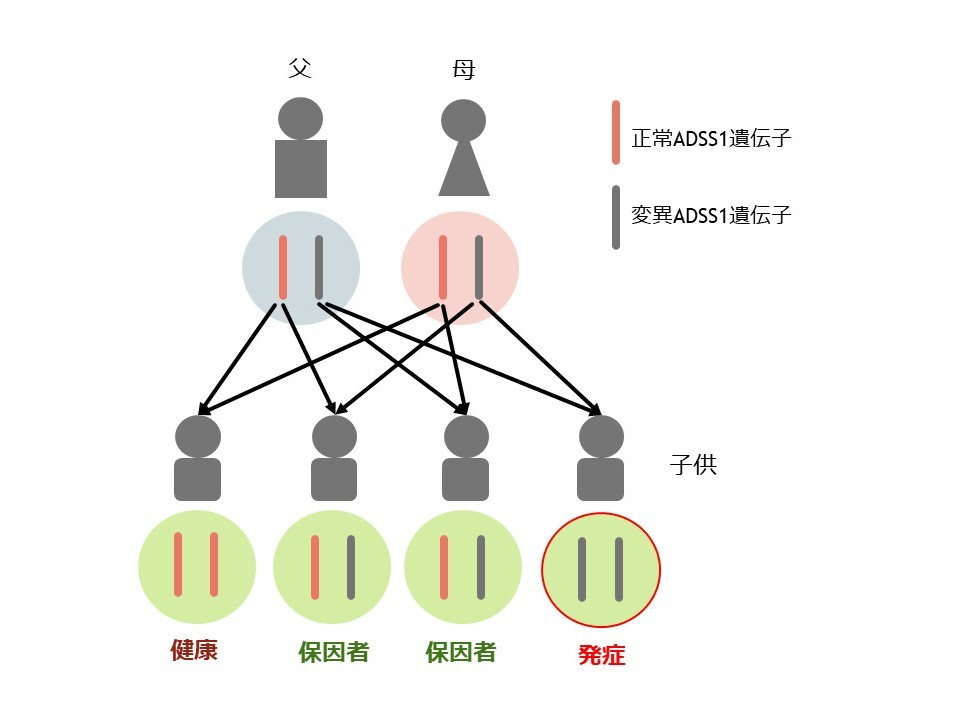
ADSS1ミオパチーは常染色体潜性遺伝(劣性遺伝)です。
病気の原因となる遺伝子が常染色体の上にあり、1対の遺伝子両方に変異があると発病する場合を潜性遺伝(劣性遺伝)と言います。
私たちは両親からそれぞれ1つずつ遺伝子を受け継ぎます。つまり、父親と母親から受け継いだ2つの遺伝子のうち、両方に変異があった場合に病気が発症します。
片方だけの変異の場合は保因者と言い、病気は発症しません。保因者の両親から両方に異常を持って生まれる確率は1/4です。
両親は23,000個もあるうちのたった1つの同じ遺伝子の1対にそれぞれ変異を持っていたことになります。
*遺伝性と言っても遺伝するわけではありません。両親から受け継いだ組み合わせが原因となります。
報告されている世界のADSS1ミオパチー患者
ADSS1遺伝子変異が遠位ミオパチー5(MPD5)の原因として、2016年に韓国から報告がありました。
その後、ADSS1ミオパチーとして、インドやトルコ、日本、アメリカからの報告はありますが、現在、診断を受けているのは世界で200人弱であり、尚且つアジア人に限定されています。
そのなかで日本人が多くを占め、およそ80人が診断されています。
ADSS1ミオパチーの治療について

ADSS1ミオパチーには現在治療薬や治療法がありません。
症状が進行していくと、最終的には歩行能力を失い、腕が動かなくなる可能性があります。
心臓、肺、嚥下筋に影響が及ぶと経管栄養や胃ろう造設、人工呼吸器のサポートが必要となる場合も出てきます。
現在、病態解明並びに治療法の研究開発が進められています。
しかし国内で診断された患者数はおよそ80名の超希少疾患なため診断できる医師は限られており、また研究資金の不足から治療法開発に大きな壁が立ちはだかっています。
治療法開発研究の現状と関連機関
ADSS1ミオパチーの研究は韓国を始め日本、アメリカでも行われています。
患者数が最も多い日本では国立精神・神経医療研究センター神経研究所で研究が行われていますが、現在資金難から研究が進まない状況です。
アメリカでは2022年に設立されたADSS1ミオパチーの患者団体CureADSSL1が支援金を募り、医学研究者や稀少疾患の専門家と協力して遺伝子治療法開発に取り組んでいます。
ADSS1ミオパチーに関する研究論文・報告等
- ADSSL1ミオパチーの多症例解析と疾患モデルマウス解析による病態の解明
https://kaken.nii.ac.jp/ja/grant/KAKENHI-PROJECT-20K16589/ - アデニルコハク酸:未開発の可能性を秘めた希少疾病用医薬品
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10304260/ - アメリカ患者団体CureADSSL1と研究機関による進捗状況
https://www.cure-adssl1.org/research.html
関連研究・医療機関
- 国立精神・神経医療研究センター神経研究所疾病研究第一部
https://www.ncnp.go.jp/nin/guide/r1/index.html - 国立精神・神経医療研究センター病院 筋疾患センター
https://www.ncnp.go.jp/hospital/guide/sd/muscular-disease.html